|
|
||
|---|---|---|
| .github/workflows | ||
| imgs | ||
| lib | ||
| notebooks | ||
| openfold | ||
| scripts | ||
| tests | ||
| CITATION.cff | ||
| Dockerfile | ||
| LICENSE | ||
| README.md | ||
| deepspeed_config.json | ||
| environment.yml | ||
| run_pretrained_openfold.py | ||
| setup.py | ||
| train_openfold.py | ||
README.md
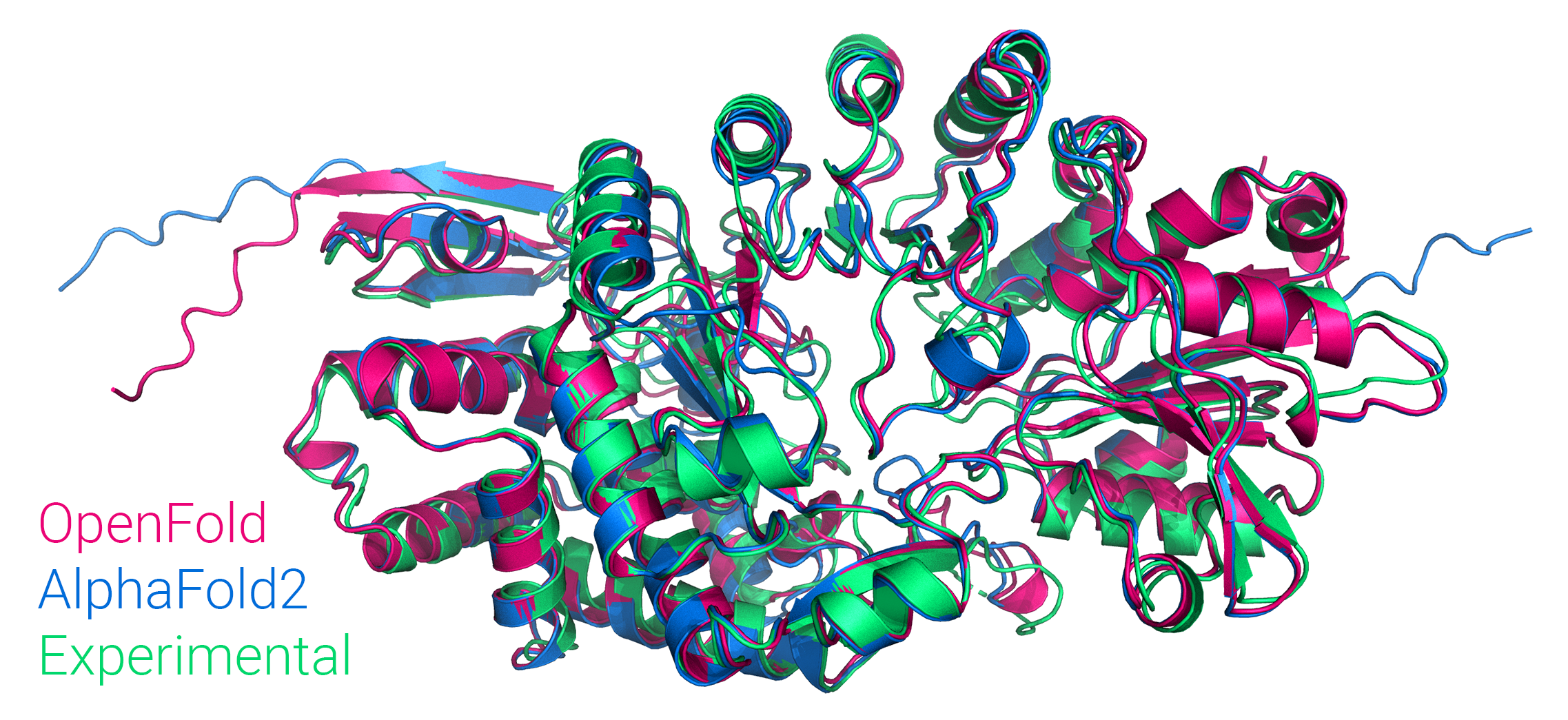
OpenFold
A faithful but trainable PyTorch reproduction of DeepMind's AlphaFold 2.
Features
OpenFold carefully reproduces (almost) all of the features of the original open source inference code (v2.0.1). The sole exception is model ensembling, which fared poorly in DeepMind's own ablation testing and is being phased out in future DeepMind experiments. It is omitted here for the sake of reducing clutter. In cases where the Nature paper differs from the source, we always defer to the latter.
OpenFold is trainable in full precision or bfloat16 with or without DeepSpeed,
and we've trained it from scratch, matching the performance of the original.
We've publicly released model weights and our training data — some 400,000
MSAs and PDB70 template hit files — under a permissive license. Model weights
are available via scripts in this repository while the MSAs are hosted by the
Registry of Open Data on AWS (RODA).
Try out running inference for yourself with our Colab notebook.
OpenFold also supports inference using AlphaFold's official parameters.
OpenFold has the following advantages over the reference implementation:
- Faster inference on GPU for chains with < 1500 residues.
- Inference on extremely long chains, made possible by our implementation of low-memory attention (Rabe & Staats 2021). OpenFold can predict the structures of sequences with more than 4000 residues on a single A100, and even longer ones with CPU offloading.
- Custom CUDA attention kernels modified from FastFold's kernels support in-place attention during inference and training. They use 4x and 5x less GPU memory than equivalent FastFold and stock PyTorch implementations, respectively.
- Efficient alignment scripts using the original AlphaFold HHblits/JackHMMER pipeline or ColabFold's, which uses the faster MMseqs2 instead. We've used them to generate millions of alignments.
Installation (Linux)
All Python dependencies are specified in environment.yml. For producing sequence
alignments, you'll also need kalign, the HH-suite,
and one of {jackhmmer, MMseqs2 (nightly build)}
installed on on your system. You'll need git-lfs to download OpenFold parameters.
Finally, some download scripts require aria2c.
For convenience, we provide a script that installs Miniconda locally, creates a
conda virtual environment, installs all Python dependencies, and downloads
useful resources, including both sets of model parameters. Run:
scripts/install_third_party_dependencies.sh
To activate the environment, run:
source scripts/activate_conda_env.sh
To deactivate it, run:
source scripts/deactivate_conda_env.sh
With the environment active, compile OpenFold's CUDA kernels with
python3 setup.py install
To install the HH-suite to /usr/bin, run
# scripts/install_hh_suite.sh
Usage
To download the databases used to train OpenFold and AlphaFold run:
bash scripts/download_data.sh data/
You have two choices for downloading protein databases, depending on whether you want to use DeepMind's MSA generation pipeline (w/ HMMR & HHblits) or ColabFold's, which uses the faster MMseqs2 instead. For the former, run:
bash scripts/download_alphafold_dbs.sh data/
For the latter, run:
bash scripts/download_mmseqs_dbs.sh data/ # downloads .tar files
bash scripts/prep_mmseqs_dbs.sh data/ # unpacks and preps the databases
Make sure to run the latter command on the machine that will be used for MSA generation (the script estimates how the precomputed database index used by MMseqs2 should be split according to the memory available on the system).
Alternatively, you can use raw MSAs from our aforementioned MSA database or
ProteinNet. After downloading
the latter database, use scripts/prep_proteinnet_msas.py to convert the data
into a format recognized by the OpenFold parser. The resulting directory
becomes the alignment_dir used in subsequent steps. Use
scripts/unpack_proteinnet.py to extract .core files from ProteinNet text
files.
For both inference and training, the model's hyperparameters can be tuned from
openfold/config.py. Of course, if you plan to perform inference using
DeepMind's pretrained parameters, you will only be able to make changes that
do not affect the shapes of model parameters. For an example of initializing
the model, consult run_pretrained_openfold.py.
Inference
To run inference on a sequence or multiple sequences using a set of DeepMind's pretrained parameters, run e.g.:
python3 run_pretrained_openfold.py \
fasta_dir \
data/pdb_mmcif/mmcif_files/ \
--uniref90_database_path data/uniref90/uniref90.fasta \
--mgnify_database_path data/mgnify/mgy_clusters_2018_12.fa \
--pdb70_database_path data/pdb70/pdb70 \
--uniclust30_database_path data/uniclust30/uniclust30_2018_08/uniclust30_2018_08 \
--output_dir ./ \
--bfd_database_path data/bfd/bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt \
--model_device "cuda:0" \
--jackhmmer_binary_path lib/conda/envs/openfold_venv/bin/jackhmmer \
--hhblits_binary_path lib/conda/envs/openfold_venv/bin/hhblits \
--hhsearch_binary_path lib/conda/envs/openfold_venv/bin/hhsearch \
--kalign_binary_path lib/conda/envs/openfold_venv/bin/kalign
--config_preset "model_1_ptm"
--openfold_checkpoint_path openfold/resources/openfold_params/finetuning_2_ptm.pt
where data is the same directory as in the previous step. If jackhmmer,
hhblits, hhsearch and kalign are available at the default path of
/usr/bin, their binary_path command-line arguments can be dropped.
If you've already computed alignments for the query, you have the option to
skip the expensive alignment computation here with
--use_precomputed_alignments.
Exactly one of --openfold_checkpoint_path or --jax_param_path must be specified
to run the inference script. These accept .pt/DeepSpeed OpenFold checkpoints
and AlphaFold's .npz JAX parameter files, respectively. For a breakdown of the
differences between the different parameter files, see the README downloaded to
openfold/resources/openfold_params/. Since OpenFold was trained under a
newer training schedule than the one from which the model_n config
presets are derived, there is no clean correspondence between config_preset
settings and OpenFold checkpoints; the only restraint is that *_ptm
checkpoints must be run with *_ptm config presets.
Note that chunking (as defined in section 1.11.8 of the AlphaFold 2 supplement)
is enabled by default in inference mode. To disable it, set globals.chunk_size
to None in the config. If a value is specified, OpenFold will attempt to
dynamically tune it, considering the chunk size specified in the config as a
minimum. This tuning process automatically ensures consistently fast runtimes
regardless of input sequence length, but it also introduces some runtime
variability, which may be undesirable for certain users. It is also recommended
to disable this feature for very long chains (see below). To do so, set the
tune_chunk_size option in the config to False.
Input FASTA files containing multiple sequences are treated as complexes. In
this case, the inference script runs AlphaFold-Gap, a hack proposed
here, using
the specified stock AlphaFold/OpenFold parameters (NOT AlphaFold-Multimer). To
run inference with AlphaFold-Multimer, use the (experimental) multimer branch
instead.
To minimize memory usage during inference on long sequences, consider the following changes:
- As noted in the AlphaFold-Multimer paper, the AlphaFold/OpenFold template
stack is a major memory bottleneck for inference on long sequences. OpenFold
supports two mutually exclusive inference modes to address this issue. One,
average_templatesin thetemplatesection of the config, is similar to the solution offered by AlphaFold-Multimer, which is simply to average individual template representations. Our version is modified slightly to accommodate weights trained using the standard template algorithm. Using said weights, we notice no significant difference in performance between our averaged template embeddings and the standard ones. The second,offload_templates, temporarily offloads individual template embeddings into CPU memory. The former is an approximation while the latter is slightly slower; both are memory-efficient and allow the model to utilize arbitrarily many templates across sequence lengths. Both are disabled by default, and it is up to the user to determine which best suits their needs, if either. - Inference-time low-memory attention (LMA) can be enabled in the model config.
This setting trades off speed for vastly improved memory usage. By default,
LMA is run with query and key chunk sizes of 1024 and 4096, respectively.
These represent a favorable tradeoff in most memory-constrained cases.
Powerusers can choose to tweak these settings in
openfold/model/primitives.py. For more information on the LMA algorithm, see the aforementioned Staats & Rabe preprint. - Disable
tune_chunk_sizefor long sequences. Past a certain point, it only wastes time. - As a last resort, consider enabling
offload_inference. This enables more extensive CPU offloading at various bottlenecks throughout the model.
Using the most conservative settings, we were able to run inference on a 4600-residue complex with a single A100. Compared to AlphaFold's own memory offloading mode, ours is considerably faster; the same complex takes the more efficent AlphaFold-Multimer more than double the time.
Training
To train the model, you will first need to precompute protein alignments.
You have two options. You can use the same procedure DeepMind used by running the following:
python3 scripts/precompute_alignments.py mmcif_dir/ alignment_dir/ \
--uniref90_database_path data/uniref90/uniref90.fasta \
--mgnify_database_path data/mgnify/mgy_clusters_2018_12.fa \
--pdb70_database_path data/pdb70/pdb70 \
--uniclust30_database_path data/uniclust30/uniclust30_2018_08/uniclust30_2018_08 \
--bfd_database_path data/bfd/bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt \
--cpus 16 \
--jackhmmer_binary_path lib/conda/envs/openfold_venv/bin/jackhmmer \
--hhblits_binary_path lib/conda/envs/openfold_venv/bin/hhblits \
--hhsearch_binary_path lib/conda/envs/openfold_venv/bin/hhsearch \
--kalign_binary_path lib/conda/envs/openfold_venv/bin/kalign
As noted before, you can skip the binary_path arguments if these binaries are
at /usr/bin. Expect this step to take a very long time, even for small
numbers of proteins.
Alternatively, you can generate MSAs with the ColabFold pipeline (and templates with HHsearch) with:
python3 scripts/precompute_alignments_mmseqs.py input.fasta \
data/mmseqs_dbs \
uniref30_2103_db \
alignment_dir \
~/MMseqs2/build/bin/mmseqs \
/usr/bin/hhsearch \
--env_db colabfold_envdb_202108_db
--pdb70 data/pdb70/pdb70
where input.fasta is a FASTA file containing one or more query sequences. To
generate an input FASTA from a directory of mmCIF and/or ProteinNet .core
files, we provide scripts/data_dir_to_fasta.py.
Next, generate a cache of certain datapoints in the template mmCIF files:
python3 scripts/generate_mmcif_cache.py \
mmcif_dir/ \
mmcif_cache.json \
--no_workers 16
This cache is used to pre-filter templates.
Next, generate a separate chain-level cache with data used for training-time data filtering:
python3 scripts/generate_chain_data_cache.py \
mmcif_dir/ \
chain_data_cache.json \
--cluster_file clusters-by-entity-40.txt \
--no_workers 16
where the cluster_file argument is a file of chain clusters, one cluster
per line (e.g. PDB40).
Finally, call the training script:
python3 train_openfold.py mmcif_dir/ alignment_dir/ template_mmcif_dir/ \
2021-10-10 \
--template_release_dates_cache_path mmcif_cache.json \
--precision 16 \
--gpus 8 --replace_sampler_ddp=True \
--seed 42 \ # in multi-gpu settings, the seed must be specified
--deepspeed_config_path deepspeed_config.json \
--checkpoint_every_epoch \
--resume_from_ckpt ckpt_dir/ \
--train_chain_data_cache_path chain_data_cache.json
where --template_release_dates_cache_path is a path to the mmCIF cache.
Note that template_mmcif_dir can be the same as mmcif_dir which contains
training targets. A suitable DeepSpeed configuration file can be generated with
scripts/build_deepspeed_config.py. The training script is
written with PyTorch Lightning
and supports the full range of training options that entails, including
multi-node distributed training, validation, and so on. For more information,
consult PyTorch Lightning documentation and the --help flag of the training
script.
If you're using your own MSAs or MSAs from the RODA repository, make sure that
the alignment_dir contains one directory per chain and that each of these
contains alignments (.sto, .a3m, and .hhr) corresponding to that chain.
Note that, despite its variable name, mmcif_dir can also contain PDB files
or even ProteinNet .core files. To emulate the AlphaFold training procedure,
which uses a self-distillation set subject to special preprocessing steps, use
the family of --distillation flags.
Testing
To run unit tests, use
scripts/run_unit_tests.sh
The script is a thin wrapper around Python's unittest suite, and recognizes
unittest arguments. E.g., to run a specific test verbosely:
scripts/run_unit_tests.sh -v tests.test_model
Certain tests require that AlphaFold (v2.0.1) be installed in the same Python
environment. These run components of AlphaFold and OpenFold side by side and
ensure that output activations are adequately similar. For most modules, we
target a maximum pointwise difference of 1e-4.
Building and using the docker container
Building the docker image
Openfold can be built as a docker container using the included dockerfile. To build it, run the following command from the root of this repository:
docker build -t openfold .
Running the docker container
The built container contains both run_pretrained_openfold.py and train_openfold.py as well as all necessary software dependencies. It does not contain the model parameters, sequence, or structural databases. These should be downloaded to the host machine following the instructions in the Usage section above.
The docker container installs all conda components to the base conda environment in /opt/conda, and installs openfold itself in /opt/openfold,
Before running the docker container, you can verify that your docker installation is able to properly communicate with your GPU by running the following command:
docker run --rm --gpus all nvidia/cuda:11.0-base nvidia-smi
Note the --gpus all option passed to docker run. This option is necessary in order for the container to use the GPUs on the host machine.
To run the inference code under docker, you can use a command like the one below. In this example, parameters and sequences from the alphafold dataset are being used and are located at /mnt/alphafold_database on the host machine, and the input files are located in the current working directory. You can adjust the volume mount locations as needed to reflect the locations of your data.
docker run \
--gpus all \
-v $PWD/:/data \
-v /mnt/alphafold_database/:/database \
-ti openfold:latest \
python3 /opt/openfold/run_pretrained_openfold.py \
/data/fasta_dir \
/database/pdb_mmcif/mmcif_files/ \
--uniref90_database_path /database/uniref90/uniref90.fasta \
--mgnify_database_path /database/mgnify/mgy_clusters_2018_12.fa \
--pdb70_database_path /database/pdb70/pdb70 \
--uniclust30_database_path /database/uniclust30/uniclust30_2018_08/uniclust30_2018_08 \
--output_dir /data \
--bfd_database_path /database/bfd/bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt \
--model_device cuda:0 \
--jackhmmer_binary_path /opt/conda/bin/jackhmmer \
--hhblits_binary_path /opt/conda/bin/hhblits \
--hhsearch_binary_path /opt/conda/bin/hhsearch \
--kalign_binary_path /opt/conda/bin/kalign \
--openfold_checkpoint_path /database/openfold_params/finetuning_2_ptm.pt
Copyright notice
While AlphaFold's and, by extension, OpenFold's source code is licensed under
the permissive Apache Licence, Version 2.0, DeepMind's pretrained parameters
fall under the CC BY 4.0 license, a copy of which is downloaded to
openfold/resources/params by the installation script. Note that the latter
replaces the original, more restrictive CC BY-NC 4.0 license as of January 2022.
Contributing
If you encounter problems using OpenFold, feel free to create an issue! We also welcome pull requests from the community.
Citing this work
For now, cite OpenFold as follows:
@software{Ahdritz_OpenFold_2021,
author = {Ahdritz, Gustaf and Bouatta, Nazim and Kadyan, Sachin and Xia, Qinghui and Gerecke, William and AlQuraishi, Mohammed},
doi = {10.5281/zenodo.5709539},
month = {11},
title = {{OpenFold}},
url = {https://github.com/aqlaboratory/openfold},
year = {2021}
}
Any work that cites OpenFold should also cite AlphaFold.